
[ad_1]
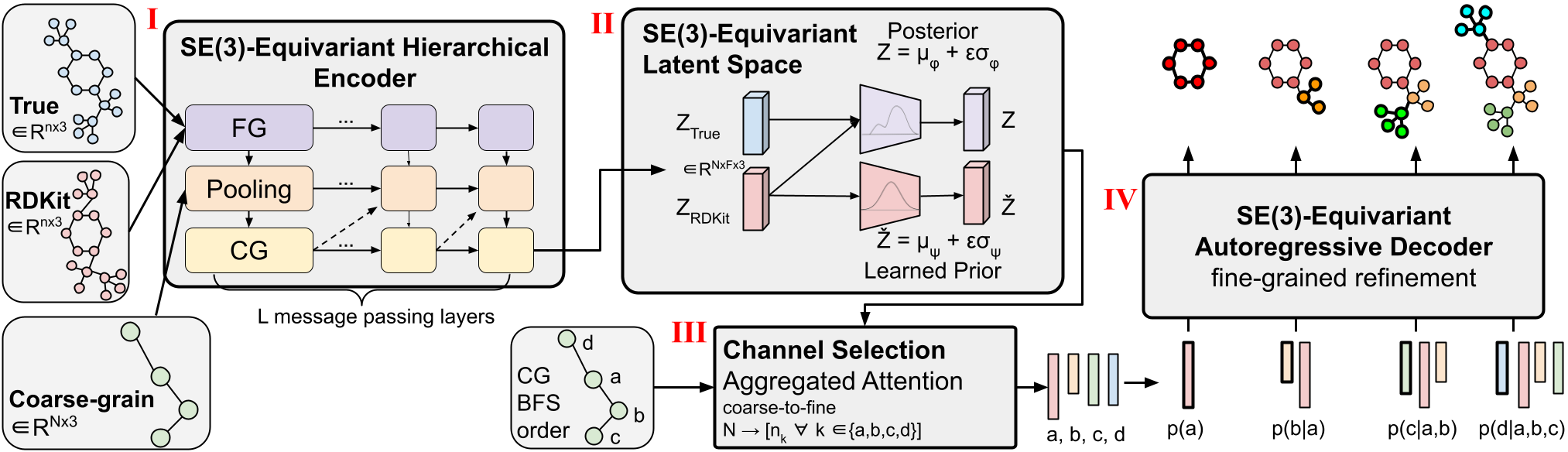
Figura 1: Arquitectura de CoarsenConf.
La generación de confórmeros moleculares es una tarea fundamental en química computacional. El objetivo es utilizar la molécula 2D para predecir estructuras moleculares 3D estables y de baja energía, los llamados confórmeros. Las conformaciones moleculares precisas son fundamentales para diversas aplicaciones que dependen de cualidades espaciales y geométricas precisas, incluido el descubrimiento de fármacos y el acoplamiento de proteínas.
Presentamos CoarsenConf, un codificador automático variacional jerárquico (VAE) equivariante SE(3) que sintetiza información a partir de coordenadas atómicas de grano fino en una representación de nivel de subgrafo de grano grueso para permitir una generación eficiente de conformadores autorregresivos.
fondo
El análisis de grano grueso reduce la dimensionalidad del problema y permite la generación autorregresiva condicional, en lugar de generar todas las coordenadas de forma independiente, como se hizo en trabajos anteriores. Al condicionar directamente las coordenadas 3D de los subgrafos generados previamente, nuestro modelo se puede generalizar mejor a subgrafos química y espacialmente similares. Esto imita el proceso de síntesis molecular subyacente en el que pequeñas unidades funcionales se combinan para formar grandes moléculas similares a fármacos. A diferencia de los métodos anteriores, CoarsenConf genera conformadores de baja energía y brinda la capacidad de modelar directamente coordenadas atómicas, distancias y ángulos de torsión.
La arquitectura CoarsenConf se puede dividir en los siguientes componentes:
(I) El codificador $q_\phi(z| ) conforma $\mathcal{C}$ como entradas (derivadas de $X$ y una estrategia CG predefinida) y genera una representación CG equivariante de longitud variable a través de mensajes equivariantes y convoluciones puntuales.
(II) Se aplican MLP equivalentes para encontrar la media y la varianza logarítmica de las distribuciones anterior y posterior.
(iii) El posterior (entrenamiento) o el previo (inferencia) se muestrea y se introduce en el módulo de selección de canales, donde se utiliza una capa de atención para aprender la ruta óptima desde el CG a la estructura FG.
(IV) Dado el vector FG latente y la aproximación RDKit, el decodificador $p_\theta(X |\mathcal{R}, z)$ aprende a recuperar la estructura FG de baja energía mediante mensajes equivariantes autorregresivos. Todo el modelo se puede entrenar de un extremo a otro optimizando la divergencia KL de las distribuciones latentes y el error de reconstrucción de los confórmeros generados.
Formalismo de tareas MCG
Formalizamos la tarea de generación de conformadores moleculares (MCG) como modelando la distribución condicional $p(X|\mathcal{R})$, donde $\mathcal{R}$ es el conformador aproximado generado por RDKit y $X$ es el óptimo. conformador (e) con baja energía. RDKit, una biblioteca de quimioinformática de uso común, utiliza un algoritmo basado en geometría de distancia de bajo costo seguido de una optimización basada en física de bajo costo para lograr aproximaciones de conformadores razonables.
De grano grueso

Figura 2: Proceso de grano grueso.
(I) Ejemplo de grano grueso de longitud variable. Las moléculas de grano fino se escinden a lo largo de enlaces giratorios que definen ángulos de torsión. Luego se vuelven de grano grueso para reducir la dimensionalidad y aprender una distribución latente a nivel de subgrafo. (II) Visualización de un conformador 3D. Se resaltan pares de átomos específicos para las operaciones de mensajería del decodificador.
El grosor molecular simplifica la representación de una molécula al agrupar los átomos de grano fino (FG) en la estructura original en perlas individuales de grano grueso (CG) $\mathcal{B}$ usando un mapeo basado en reglas, como se muestra en la Figura 2. (I). La granulación gruesa se usa ampliamente en el diseño molecular y de proteínas y, de manera similar, la generación de niveles de fragmentos o subgrafos ha demostrado ser extremadamente valiosa en diversas tareas de diseño molecular 2D. Descomponer problemas generativos en partes más pequeñas es un enfoque que se puede aplicar a múltiples tareas moleculares 3D y proporciona una reducción de dimensionalidad natural para permitir trabajar con sistemas grandes y complejos.
Observamos que, en comparación con trabajos anteriores que se centran en estrategias de CG de longitud fija, donde cada molécula se representa con una resolución fija de $N$ perlas de CG, nuestro método admite CG de longitud variable debido a su flexibilidad y capacidad utiliza cualquier opción de grosor. técnica de arena. Esto significa que un único modelo CoarsenConf se puede generalizar a cualquier resolución de grano grueso porque las moléculas de entrada se pueden asignar a cualquier número de perlas CG. En nuestro caso, los átomos formados por cada componente enlazado formado por la separación de todos los enlaces giratorios se vuelven gruesos en una sola perla. Esta elección en el método CG obliga implícitamente al modelo a aprender sobre los ángulos de torsión, así como sobre las coordenadas atómicas y las distancias interatómicas. En nuestros experimentos, utilizamos GEOM-QM9 y GEOM-DRUGS, que tienen en promedio 11 átomos y 3 perlas CG y 44 átomos y 9 perlas CG, respectivamente.
Equivarianza SE(3)
Un aspecto importante del trabajo con estructuras 3D es mantener la equivarianza adecuada. Las moléculas tridimensionales son equivalentes bajo rotaciones y traslaciones, o equivarianza SE(3). Aplicamos la equivarianza SE(3) en el codificador, decodificador y espacio latente de nuestro modelo probabilístico CoarsenConf. Como resultado, $p(X | \mathcal {R})$ permanece sin cambios para cada rototraducción del confórmero aproximado $\mathcal {R}$. Además, si $\mathcal{R}$ se gira 90° en el sentido de las agujas del reloj, esperamos que el $X$ óptimo tenga la misma rotación. Para obtener una definición detallada y una discusión de los métodos para mantener la equivarianza, consulte el documento completo.
Atención agregada
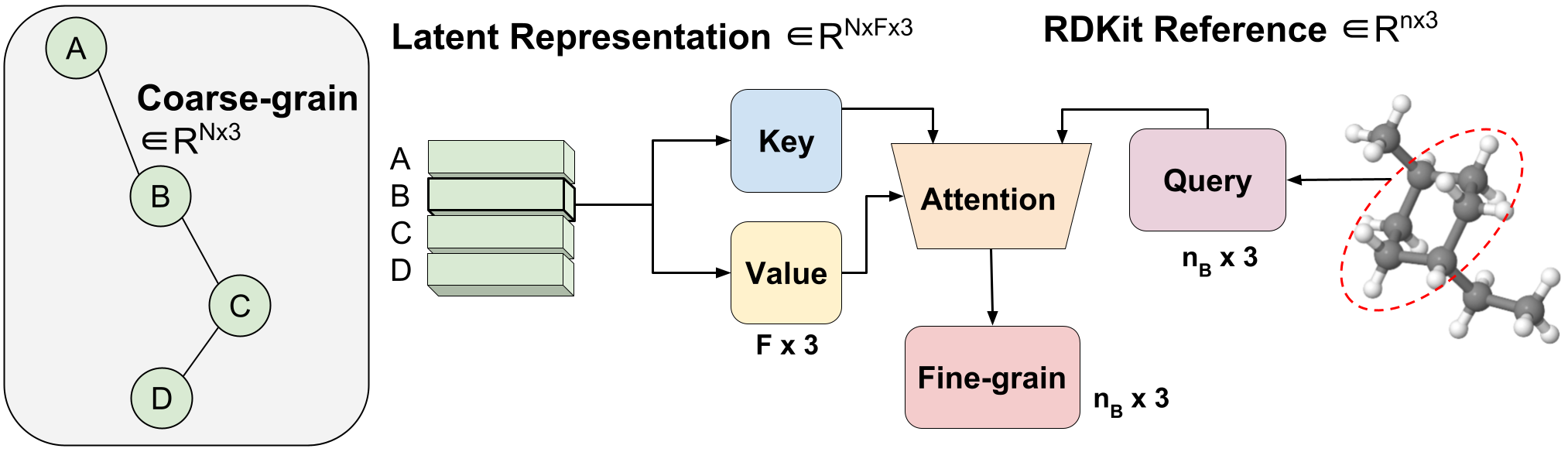
Figura 3: Backmapping de longitud variable de grueso a fino mediante Atención Agregada.
Introducimos un método, al que llamamos Atención Agregada, para aprender el mapeo óptimo de longitud variable desde la representación latente CG a las coordenadas FG. Esta es una operación de longitud variable porque una sola molécula con $n$ átomos se puede asignar a cualquier número de $N$ perlas CG (cada perla está representada por un único vector latente). El vector latente de una única cuenta CG $Z_{B}$ $\in R^{F \times 3}$ se utiliza como clave y valor de una única operación de atención de cabeza con una dimensión de incrustación de tres para que coincida con x. Coordenadas y, z. El vector de consulta es el subconjunto del conformador RDKit correspondiente a la cuenta $B$ $\in R^{ n_{B} \times 3}$, donde $n_B$ es de longitud variable ya que sabemos a priori cuántos átomos de FG corresponden a una cuenta CG específica. Al utilizar la atención, aprendemos de manera eficiente la combinación óptima de características latentes para la reconstrucción de FG. A esto lo llamamos atención agregada porque agrega segmentos 3D de información FG para formar nuestra consulta latente. La Atención Agregada es responsable de la traducción eficiente de la representación CG latente a coordenadas FG utilizables (Figura 1 (III)).
Modelo
CoarsenConf es un VAE jerárquico con un codificador y decodificador equivalente a SE(3). El codificador funciona con características del átomo invariante SE(3) $h \in R^{ n \times D}$ y coordenadas del átomo equivariante SE(3) $x \in R^{n \times 3}$. Una única capa de codificador consta de tres módulos: de grano fino, de agrupación y de grano grueso. Las ecuaciones completas de cada módulo se pueden encontrar en el documento completo. El codificador produce un tensor CG equivariante final $Z \in R^{N \times F \times 3}$, donde $N$ es el número de cuentas y F es el tamaño latente definido por el usuario.
El decodificador tiene dos tareas. La primera es transformar la representación latente y tosca nuevamente al espacio FG a través de un proceso que llamamos selección de canal, que aprovecha la atención agregada. El segundo es refinar autorregresivamente la representación detallada para generar las coordenadas finales de baja energía (Figura 1 (IV)).
Enfatizamos que nuestro modelo aprende los ángulos de torsión óptimos de manera no supervisada mediante conectividad de ángulo de torsión de grano grueso, ya que la entrada condicional al decodificador no está alineada. CoarsenConf garantiza que cada subgrafo generado a continuación se gire correctamente para lograr un error de distancia y coordenadas bajo.
Resultados experimentales
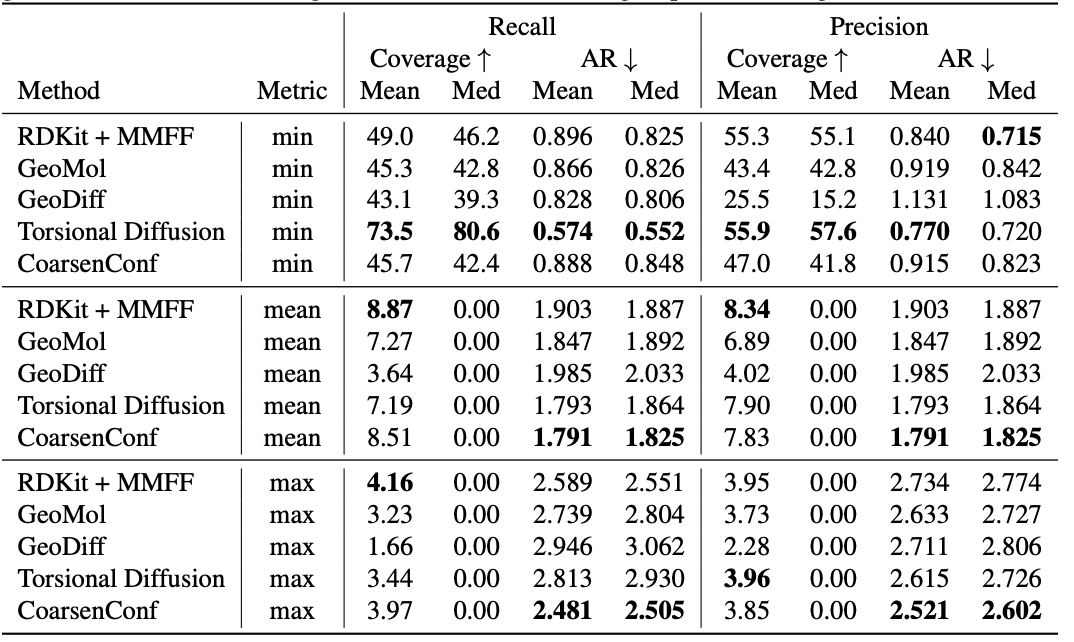
tabla 1: Calidad de los conjuntos de conformadores generados para el conjunto de prueba GEOM-DRUGS ($\delta=0,75 Å$) en términos de cobertura (%) y RMSD promedio ($Å$). CoarsenConf (5 épocas) se limitó a utilizar el 7,3% de los datos utilizados por Torsional Diffusion (250 épocas) para ilustrar un bajo costo computacional y un régimen de restricción de datos.
El error promedio (AR) es la métrica clave que mide el RMSD promedio para las moléculas generadas del conjunto de prueba correspondiente. La cobertura mide el porcentaje de moléculas que se pueden generar dentro de un umbral de error determinado ($\delta$). Introducimos las métricas Media y Máx para evaluar mejor la generación robusta y evitar el sesgo de muestreo de la métrica Mín. Destacamos que la métrica Min proporciona resultados intangibles ya que no hay forma de saber cuál de los confórmeros generados por 2L es el mejor para una sola molécula a menos que se conozca a priori el confórmero óptimo. La Tabla 1 muestra que CoarsenConf produce el promedio más bajo y el peor error en todo el conjunto de prueba de moléculas de DROGAS. Además, mostramos que RDKit logra una mejor cobertura que la mayoría de los métodos basados en aprendizaje profundo utilizando optimización basada en física de bajo costo (MMFF). Para definiciones formales de las métricas y discusión adicional, consulte el documento completo vinculado a continuación.
Para obtener más información sobre CoarsenConf, consulte el artículo sobre arXiv.
BibTex
Si CoarsenConf inspira su trabajo, considere citarlo con:
@article{reidenbach2023coarsenconf,
title={CoarsenConf: Equivariant Coarsening with Aggregated Attention for Molecular Conformer Generation},
author={Danny Reidenbach and Aditi S. Krishnapriyan},
journal={arXiv preprint arXiv:2306.14852},
year={2023},
}
[ad_2]