
[ad_1]
(Noticias de Nanowerk) Una de las muchas peculiaridades de la mecánica cuántica es que las partículas también pueden describirse como ondas. Un ejemplo común es el fotón, la partícula asociada con la luz.
En estructuras ordenadas, los llamados cristales, los electrones se pueden ver y describir como ondas que se propagan por todo el sistema, una imagen bastante armoniosa. A medida que los electrones se mueven a través del cristal, los iones (átomos que llevan una carga negativa o positiva) se organizan periódicamente en el espacio.
Ahora bien, si tuviéramos que agregar un electrón adicional al cristal, su carga negativa podría hacer que los iones a su alrededor se alejen de su posición de equilibrio. La carga del electrón se localizaría en el espacio y se acoplaría a las distorsiones estructurales circundantes – «red» – del cristal, creando una nueva partícula conocida como polarón.
«Técnicamente, un polarón es una cuasipartícula que consiste en un electrón que es ‘atraído’ por sus fonones autoinducidos, que representan las vibraciones cuantificadas del cristal», dice Stefano Falletta de la Escuela de Ciencias Básicas de la EPFL. Continúa: “La estabilidad de Polaron surge de una competencia entre dos contribuciones de energía: la ganancia debido a la localización de la carga y el costo debido a las distorsiones de la red. A medida que el polarón se desestabiliza, el electrón adicional se deslocaliza por todo el sistema mientras los iones recuperan sus posiciones de equilibrio”.
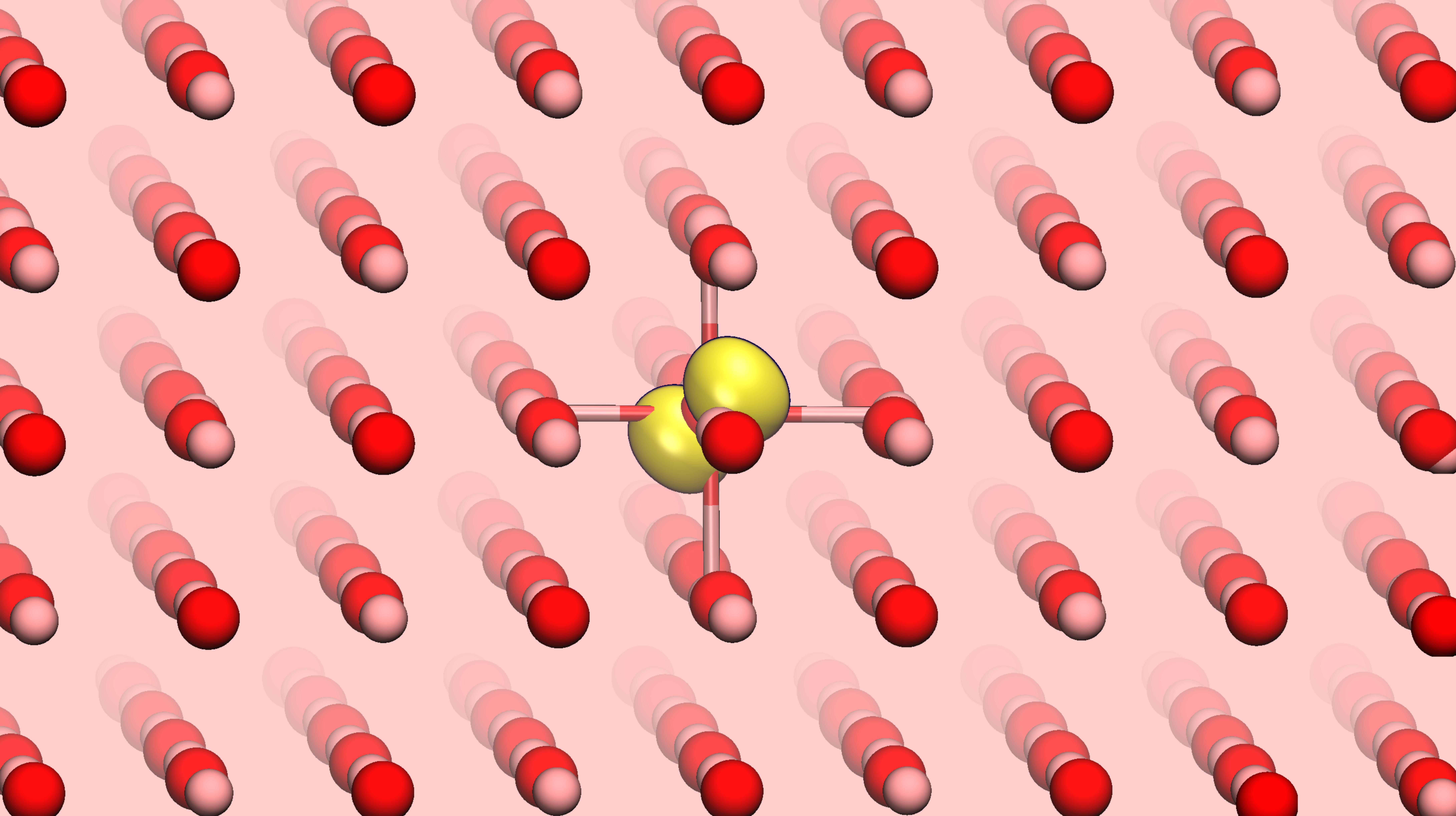
En colaboración con el profesor Alfredo Pasquarello de la EPFL, han publicado dos artículos en Cartas de verificación física («Autointeracción de muchos cuerpos y polarones») y Comprobación física B («Polarones libres de la autointeracción de muchos cuerpos en la teoría funcional de la densidad») describe un nuevo enfoque para resolver un defecto importante en una teoría establecida que los físicos usan para estudiar las interacciones de los electrones en los materiales. El método se denomina teoría funcional de la densidad o DFT y se utiliza en física, química y ciencia de los materiales para estudiar la estructura electrónica de sistemas de muchas partículas, como átomos y moléculas.
DFT es una poderosa herramienta para realizar cálculos ab initio en materiales al simplificar el tratamiento de las interacciones de electrones. Sin embargo, DFT es propenso a perturbar las interacciones del electrón consigo mismo, lo que los físicos llaman el «problema de la autointeracción». Esta autointeracción es una de las principales limitaciones de DFT y, a menudo, conduce a una descripción incorrecta de los polarones, que a menudo están desestabilizados.
«En nuestro trabajo, presentamos una formulación teórica para la interacción electrón-yo que resuelve el problema de la localización del polarón en la teoría funcional de la densidad», dice Falletta. “Esto permite el acceso a estabilidades precisas de polarones dentro de un esquema computacionalmente eficiente. Nuestro estudio allana el camino hacia cálculos sin precedentes de polarones en grandes sistemas, en estudios sistemáticos que involucran grandes cantidades de material o en dinámicas moleculares que evolucionan durante largos períodos de tiempo”.
[ad_2]