
[ad_1]
(Foco Nanowerk) La búsqueda de una electrónica orgánica más eficiente, desde pantallas OLED hasta células solares orgánicas, es un desafío duradero en toda la ciencia de los materiales. Pero la amplia variedad de posibles compuestos orgánicos y las limitaciones de los métodos de detección virtual (técnicas computacionales avanzadas para evaluar y predecir las propiedades de los materiales) han obstaculizado el ritmo del descubrimiento de nuevos materiales.
Los investigadores han intentado navegar sistemáticamente en el espacio químico y predecir con precisión las propiedades moleculares para descubrir nuevos compuestos potentes. Sin embargo, los enfoques existentes tienden a centrar la investigación en mejoras incrementales o les resulta difícil extraer principios de diseño generativo.
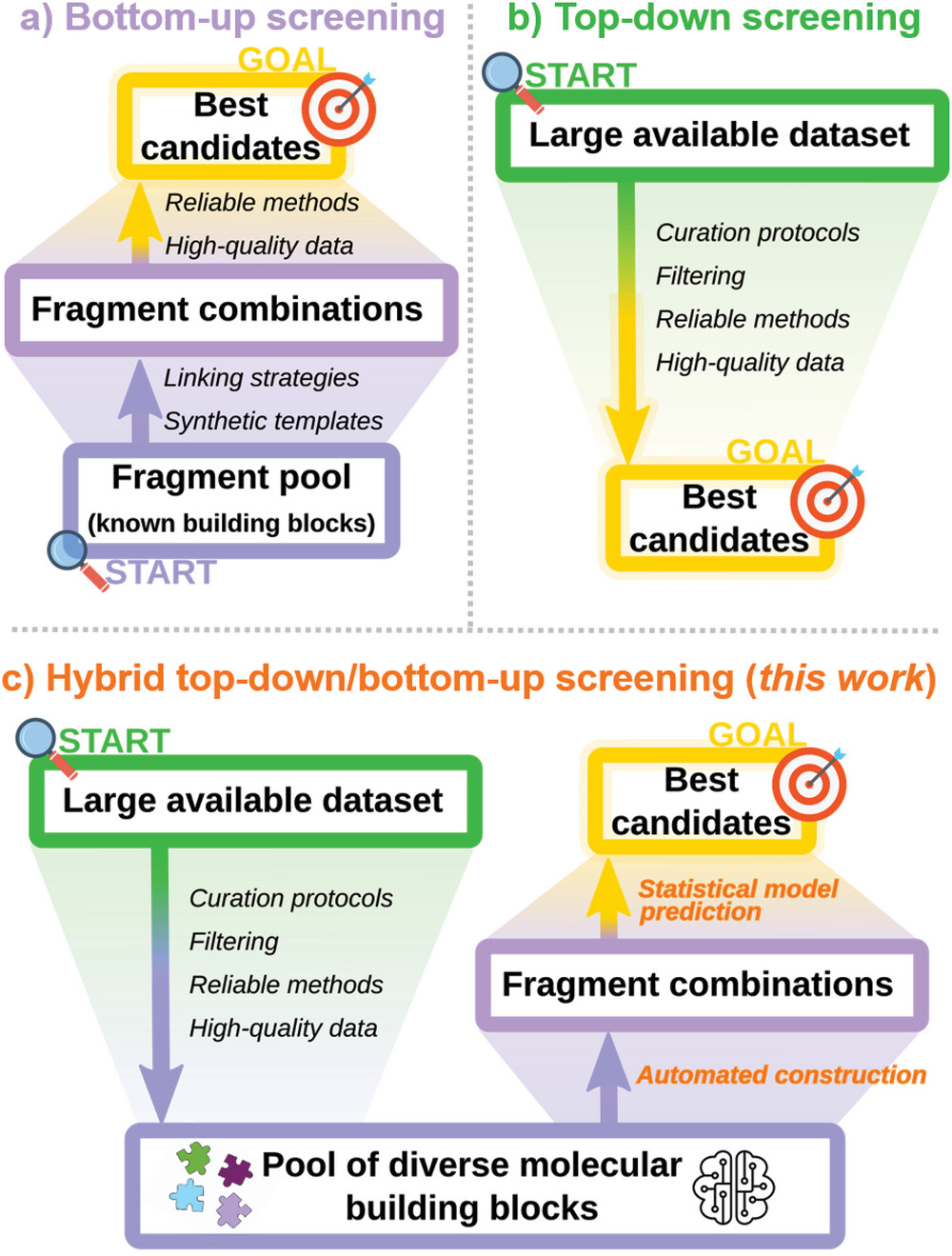
Por un lado, las estrategias ascendentes comienzan con componentes moleculares conocidos y los combinan de diferentes maneras, lo que permite una detección virtual rápida. Sin embargo, se centran en mejoras incrementales de los materiales existentes en lugar de familias de compuestos completamente nuevas. Por otro lado, la minería de datos descendentes de bases de datos estructurales arroja una red más amplia pero tiene dificultades para extraer pautas de diseño viables a partir de los resultados.
En el campo de la ciencia de materiales, existen dos estrategias principales para descubrir nuevos materiales. Por un lado, las estrategias “de abajo hacia arriba” comienzan con componentes moleculares conocidos. Piense en ello como crear nuevas estructuras con ladrillos LEGO. Los científicos combinan estos bloques de varias maneras, lo que permite realizar pruebas rápidas de sus propiedades por computadora. Sin embargo, este método se centra en pequeñas mejoras de los materiales existentes en lugar de crear materiales completamente nuevos. Por otro lado, la minería de datos “de arriba hacia abajo” implica buscar en enormes bases de datos de estructuras moleculares para encontrar nuevos materiales potenciales. Es como lanzar una amplia red para ver qué se puede encontrar, pero este enfoque a menudo tiene dificultades para identificar pautas prácticas para crear estos materiales a partir de los datos descubiertos.
Al mismo tiempo, los altos costos computacionales impiden que ambos enfoques exploren espacios moleculares tan grandes como la propia naturaleza. Los modelos predictivos de aprendizaje automático han surgido como una forma de evitar costosos cálculos químicos cuánticos. Sin embargo, el desarrollo de modelos se ve obstaculizado por la falta de conjuntos de datos de entrenamiento de alta calidad y a gran escala con propiedades relevantes del estado excitado calculadas a partir de los primeros principios.
Ahora, los investigadores de la EPFL han combinado la ingeniería ascendente con la extracción de datos de arriba hacia abajo en una plataforma híbrida de descubrimiento de materiales. Los avances innovadores son un conjunto de datos curado de 117.000 estructuras orgánicas sintetizadas con propiedades calculadas por TDDFT, identificación automatizada de sitios de acoplamiento cruzado para generar espontáneamente nuevos compuestos y modelos de aprendizaje automático rápidos pero precisos para predecir propiedades ópticas clave.
Los resultados fueron publicados en Materiales avanzados (“Descubrimiento basado en datos de materiales electrónicos orgánicos a través de un diseño híbrido de arriba hacia abajo y de abajo hacia arriba”).
Utilizando esta infraestructura basada en datos, los investigadores demostraron la detección a gran escala de candidatos a fisión singlete. De más de un millón de dímeros donante-aceptor construidos, se ha identificado que cientos de miles tienen energías de estado excitado ideales para células solares. Muchos presentan motivos moleculares inusuales, como nitronas y furoxanos, que rara vez se han estudiado para la fisión singlete.
El uso de toda la variedad de compuestos estables y ya sintetizados facilita la búsqueda de componentes básicos convencionales. Y al entrenar modelos con datos de precisión cuántica del espacio químico conocido, las predicciones confiables permiten la detección virtual a una escala sin precedentes.
Los investigadores extrajeron más de 167.000 estructuras cristalinas de la base de datos estructural de Cambridge, lo que representa la mayor recopilación pública de compuestos sintéticos caracterizados. Una selección exhaustiva eliminó duplicados, errores y unidades encuadernadas sin sentido.
El conjunto final filtrado de compuestos 117K proporciona una paleta estructuralmente diversa que abarca la química orgánica conocida mucho más allá de los conjuntos de datos moleculares tradicionales. Incluso en comparación con las moléculas pequeñas de 134K del conjunto QM9 establecido, esto cubre más elementos en un rango de tamaño mayor sin limitaciones.
A partir de cada estructura cristalina refinada, se calculó una variedad de propiedades electrónicas utilizando aproximaciones de funciones de densidad. Estos incluyen energías orbitales fronterizas, múltiples niveles de excitación singletes y tripletes bajos y descriptores cuantitativos del carácter de transición. Juntas, estas propiedades influyen en el comportamiento del excitón y el carácter de transferencia de carga que subyacen en las aplicaciones desde emisores OLED hasta energía fotovoltaica orgánica.
Para convertir instantáneas moleculares estáticas en bloques de construcción versátiles, los investigadores desarrollaron una herramienta basada en topología que marca sitios potenciales de carbono adecuados para reacciones de acoplamiento cruzado. Se centran en posiciones de CH insaturado y enumeran los entornos químicos locales para identificar grupos equivalentes de simetría.
Este protocolo imita un análisis retrosintético para descifrar sitios donde los fragmentos podrían ensamblarse a través de formaciones de enlaces carbono-carbono establecidas. Al codificar la capacidad de acoplamiento directo, se extrae información relevante para la síntesis de datos estructurales 3D. También permite la compilación algorítmica de derivaciones que originalmente no estaban presentes en el conjunto de datos.
Para evitar la necesidad de realizar costosos cálculos de estado excitado para cada nuevo compuesto hipotético, el equipo entrenó modelos de aprendizaje automático utilizando los datos precalculados. La representación de entrada incluía potenciales London y Axilrod-Teller Muto dependientes de la geometría: términos de pares atómicos y tripletes inspirados físicamente.
Los modelos lograron una precisión notable en la predicción de energías de excitación singlete y triplete para diversas estructuras orgánicas. Esto requirió agregar varios miles de dímeros generados por computadora a los datos de entrenamiento para representar adecuadamente los pares de moléculas unidas. Los modelos recapitulan la química cuántica con una fracción del esfuerzo computacional y predicen propiedades ópticas en un instante.
Los investigadores demostraron el poder de su infraestructura de descubrimiento híbrido construyendo más de un millón de dímeros donantes-aceptores y probando su idoneidad para la fisión singlete intramolecular. Los materiales de fisión singlete conocidos son raros, sensibles, tienen baja energía triplete y se derivan en gran medida de derivados del oligoaceno.
Al establecer criterios de diseño complementarios basados en patrones orbitales fronterizos y energías de excitación, el equipo seleccionó racionalmente fragmentos donantes y aceptores con propiedades apropiadas de su base de datos molecular. El acoplamiento automatizado de estos miles de bloques de construcción y el posterior cribado acelerado por ML produjeron casi 560.000 dímeros estructuralmente diversos con firmas termodinámicas ideales para energías de fisión singlete y triplete adecuadas para absorbentes fotovoltaicos comunes como el silicio y el CdTe.
Muchos contienen grupos aceptores rara vez estudiados, como nitronas y furoxanos, que tienen un potencial sin explotar para la fisión singlete, como lo confirman los análisis de subestructura. Partir directamente de moléculas que ya han sido sintetizadas con éxito evita la dependencia de ideas convencionales sobre subunidades químicas productivas. Esto ilustra cómo la generación de compuestos basada en datos mediante modelos predictivos proporciona acceso independiente a áreas moleculares desconocidas con propiedades deseadas.

De
Miguel
Berger
– Michael es autor de tres libros de la Royal Society of Chemistry: Nano-Society: Pushing the Boundaries of Technology, Nanotechnology: The Future is Tiny y Nanoengineering: The Skills and Tools Making Technology Invisible Copyright ©
Nanowerk LLC
¡Conviértete en autor invitado de Spotlight! Únase a nuestro gran y creciente grupo de autores invitados. ¿Acaba de publicar un artículo científico o le gustaría compartir otros desarrollos interesantes con la comunidad de nanotecnología? Cómo publicar en nanowerk.com.
[ad_2]